
Parámetros en cromatografía de gases
Tiempo de lectura estimado: 8 minutos
Hay muchos factores que afectan a la separación (y/o a los tiempos de retención) en la cromatografía de gases, como el tipo de columna, la concentración de la muestra, la temperatura del horno y el caudal del gas portador. Los factores controlables por un estudiante se describen en esta sección, y están relacionados con la concentración y la temperatura.
Dilución
Las pruebas de cromatografía deben realizarse con muestras muy diluidas. Una muestra diluida permite un equilibrio adecuado entre la fase estacionaria y la fase móvil dentro de la columna, lo que da lugar a picos estrechos de forma gaussiana. Si las muestras están demasiado concentradas, las formas de los picos suelen ser anchas y aplanadas en la parte superior (Figura 1a), lo que indica que la columna (y/o el detector) se ha saturado. Las abundancias apropiadas (el eje y en los espectros de cromatografía) utilizando un detector de espectrómetro de masas deben estar en los millones bajos (por ejemplo, 1.500.000 TICs más o menos, véase la Figura 1b).
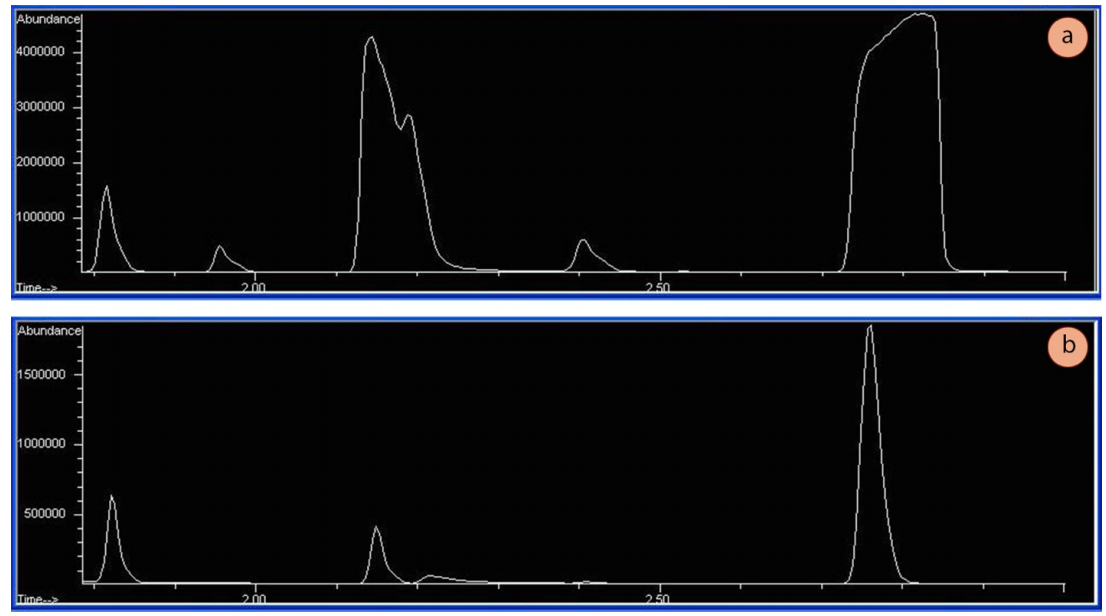
Una cromatografía de gases nunca debe funcionar con un líquido sin diluir. Las muestras demasiado concentradas no sólo hacen que los picos se amplíen y posiblemente se solapen, sino que también pueden ser perjudiciales para los detectores. Los espectrómetros de masas requieren cantidades muy pequeñas de líquido o, con el tiempo, sus filamentos se degradan.
Para preparar una muestra rutinaria de cromatografía, se necesitan 1,5mLde un disolvente de bajo punto de ebullición (por ejemplo, metanol, acetona limpia, éter dietílico o diclorometano). Se añade a un vial de cromatografía de gases (figura 2) junto con una gota de compuesto. A continuación, se inyecta aproximadamente 1μL de esta solución diluida en el equipo, que se desvía internamente, lo que hace que entren cantidades muy pequeñas en la estrecha columna capilar y el detector. El funcionamiento normal de un equipo de cromatografía requiere cantidades minúsculas de material, lo que supone una gran ventaja para esta técnica.

Retraso del disolvente (con espectrómetros de masas)
Una muestra para cromatografía correctamente preparada contiene una pequeña cantidad de compuesto disuelto en un disolvente y, sin embargo, los espectros de cromatografía de gases (cuando se emparejan con el detector de un espectrómetro de masas) no suelen mostrar ningún pico de disolvente. La razón es que los métodos de cromatografía suelen incluir un «retardo del disolvente», en el que el detector se apaga hasta que ha transcurrido un cierto tiempo después de la inyección. Incluso con la dilución dentro del equipo de GC-MS, la cantidad de disolvente que llega al detector lo sobrecargaría y acabaría por degradarlo. Por lo tanto, el detector se deja inactivo hasta que el disolvente haya pasado por la columna, y se activa después.
Si se observa cualquier espectro de cromatografía, el tiempo de retención (eje de abscisas) nunca comienza en el minuto cero, que representa el momento en que se inyecta la muestra en la columna. En el espectro de cromatografía de hexanos, el espectro comienza a los 1,40 minutos, (como se indica con una flecha en la figura 3). El disolvente habría eluido de la columna antes de ese momento.
El disolvente de la cromatografía debe elegirse de forma que el disolvente eluya antes que el compuesto de interés (el disolvente debe tener un punto de ebullición significativamente inferior al del compuesto), de forma que se pueda establecer el retardo del disolvente entre la elución del disolvente y la muestra. Por ejemplo, el metanol (p.e. 65°C) habría sido un disolvente inadecuado para la muestra de hexanos (p.e. 68°C).
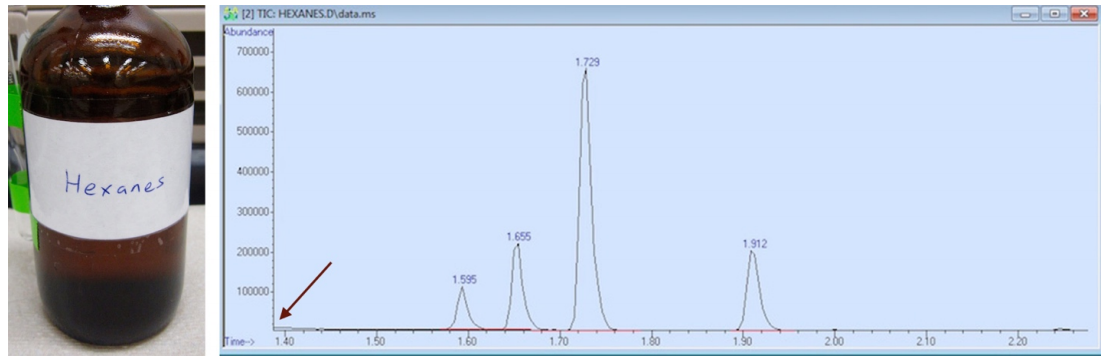
Si el retardo del disolvente se fijara de forma que se pudieran detectar las señales del hexano, el espectro de cromatografía se vería abrumado por las señales del metanol, ya que los dos compuestos tienen puntos de ebullición muy similares y eluirían bastante cerca. Si el detector se activara después de la elusión del metanol, el instrumento probablemente no detectaría los hexanos que eluyen tan cerca. Por lo tanto, se utilizó un disolvente con un punto de ebullición significativamente más bajo (éter dietílico, p.e. 35°C) para producir el espectro de cromatografía de los hexanos de la Figura 3, y este disolvente habría eluido antes del retraso de 1,40 minutos.
Temperatura del horno de cromatografía
La temperatura del horno de cromatografía es una variable fácil de ajustar. La temperatura del horno tiene un efecto dramático en los tiempos de retención, de manera muy similar a lo que el cambio de la fase móvil en TLC tiene en el Rf.
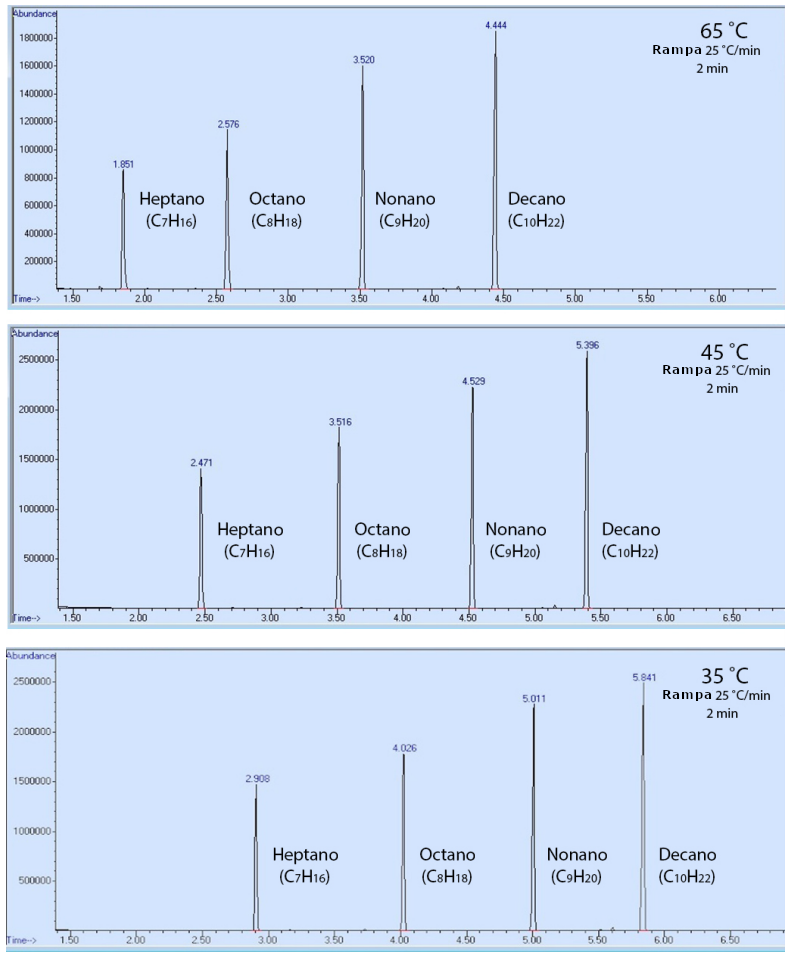
Para demostrarlo, se analizó una muestra que contenía heptano, octano, nonano y decano utilizando tres métodos de cromatografía, que sólo se diferenciaban por su temperatura inicial (65°C, 45°C o 35°C). El orden de elución de los compuestos fue el mismo en todos los métodos (Figura 4), pero los componentes eluyeron más rápidamente con una temperatura de horno más caliente (tuvieron tiempos de retención más bajos). Este resultado puede explicarse en que los compuestos fueron más capaces de superar su atracción intermolecular con la fase estacionaria (el revestimiento de la columna de cromatografía) cuando había más energía térmica disponible. Esto hizo que los compuestos pasaran menos tiempo adheridos a la fase estacionaria, y menos tiempo retenidos por la columna.
Uso de una rampa de temperatura
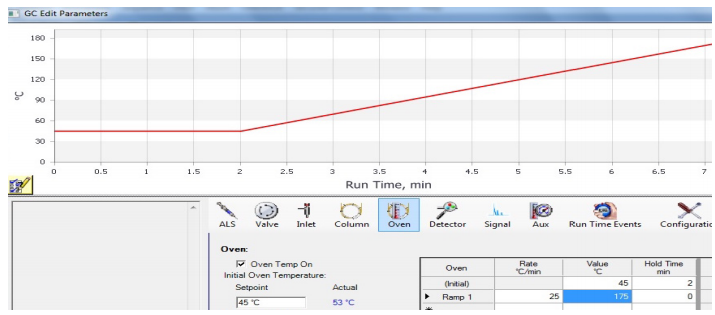
Un cromatografo puede funcionar a una temperatura de horno todo el tiempo (un funcionamiento «isotérmico»), o el método puede incluir la programación de la temperatura. En un método programado, la temperatura puede permanecer fija durante un tiempo determinado, y luego puede incluir una «rampa», en la que la temperatura se incrementa a un ritmo constante durante la ejecución. En la figura 5 se muestra un gráfico de una rampa generada por el instrumento de cromatografía.
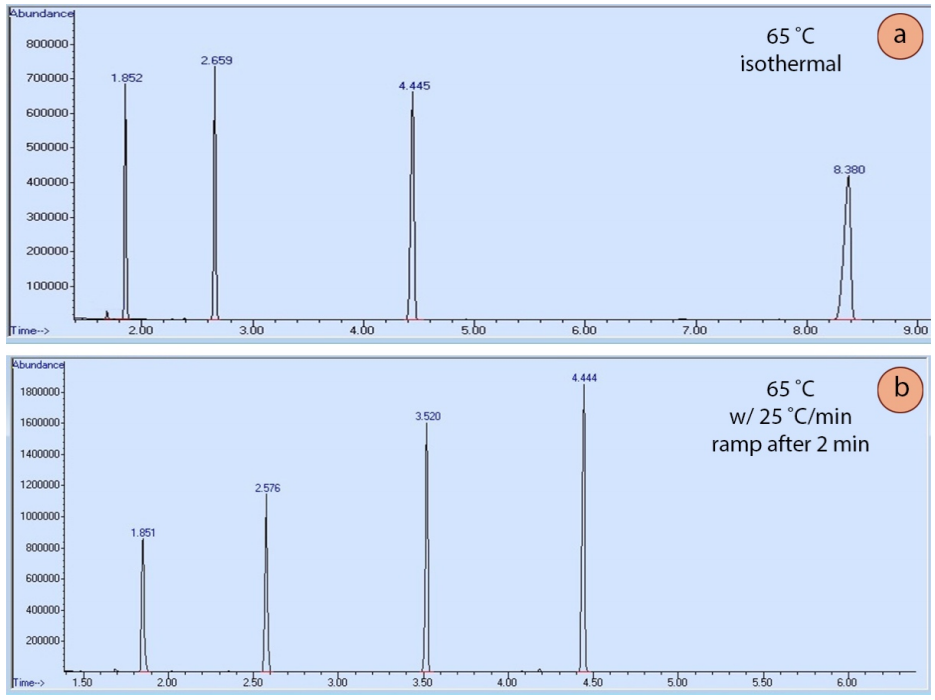
La programación de la temperatura es útil porque cuanto más tiempo permanezca un compuesto en la columna, más amplia será su elución, ya que la difusión ensancha la muestra (de forma análoga a cómo se ensanchan las manchas durante la elución en TLC). La figura 6a muestra una corrida de cromatografía isotérmica de cuatro compuestos, y la forma del pico se ensancha significativamente a medida que aumenta el tiempo de retención. La figura 6b muestra una corrida de cromatografía de los mismos cuatro compuestos eluidos usando una rampa de temperatura. La rampa permite que los compuestos eluyan más rápidamente y que la forma del pico se afine y mejore. La rampa óptima para cada situación suele requerir la experimentación para conseguir la mejor forma de pico y la mejor separación de los componentes.
Para más información GC Parameters



