
Cristalización cuantitativa
Estimated reading time: 11 minutos
En esta sección, los datos de solubilidad real de los compuestos se utilizan para describir cuantitativamente la purificación de sólidos impuros que contienen una «impureza soluble» o una impureza con solubilidad similar a la del compuesto de interés.
Cristalización cuantitativa con una «impureza soluble»
La cristalización más sencilla en términos de purificación es cuando una impureza es muy soluble en el disolvente frío mientras que el compuesto de interés no lo es (véase la secuencia de procedimiento en la figura 1). En esta situación, la impureza queda atrapada en la matriz cristalina del sólido impuro, y sólo necesita ser liberada por disolución. Después de añadir disolvente caliente para disolver el sólido impuro y luego enfriar, la impureza soluble permanece disuelta en el licor madre mientras que el compuesto de interés cristaliza y puede ser recogido por filtración de succión.
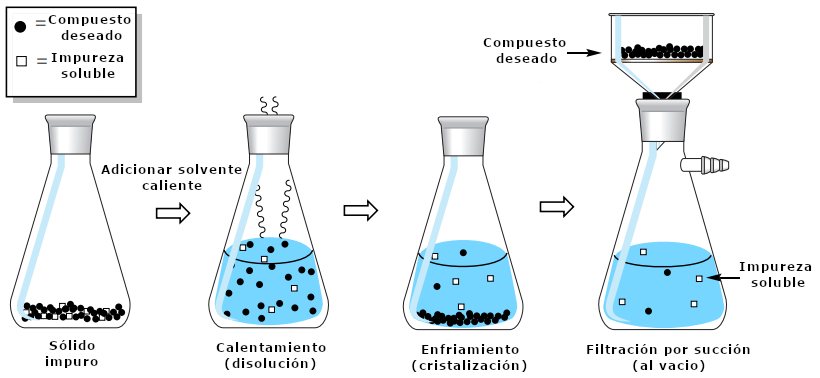
Un ejemplo de este tipo de sistema es una muestra que contiene 4,00 g de acenafteno y 0,50 g de acetanilida. Según la obra de Perrin Purification of Organic Compounds, el acenafteno puede purificarse por cristalización utilizando etanol, y aunque esta muestra contiene una cantidad considerable de impureza de acetanilida (13 mol%), los cálculos de esta sección mostrarán que este proceso debería funcionar bien.
Los valores de solubilidad del acenafteno y la acetanilida en etanol se muestran en la figura 2.
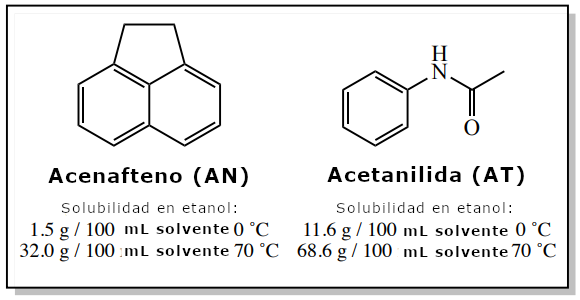
4,00 g de acenafteno puro (AN) deben disolverse completamente en 12,5 mL (ver cálculo más abajo) de etanol casi hirviendo (el punto de ebullición del etanol es de 78°C).
$$ 4.00\,\mathrm{g\ acenafteno\ (AN)} \times \frac{100\,\mathrm{mL\ etanol\ caliente}}{32.0\,\mathrm{g\ AN}} = \mathbf{12.5\,\mathrm{mL\ etanol\ caliente}} \;\left(70^\circ\mathrm{C}\right) $$Como la acetanilida (AT) es más soluble que el acenafteno a esta temperatura (véase la figura 2), también debería disolverse completamente en 12,5 mL de etanol caliente.
Si la solución etanólica caliente que contiene estos dos componentes se enfría en un baño de hielo, el acenafteno debería cristalizar, ya que tiene una baja solubilidad en el etanol frío. El siguiente cálculo muestra la cantidad de acenafteno que quedaría disuelta en el etanol frío (0,19 g). Cualquier cantidad de acenafteno presente superior a 0,19 g formaría un sólido, en este caso 3,81 g (4,00 g-0,19 g).
$$ 12.5\,\mathrm{mL\ etanol\ frío} \times \frac{1.5\,\mathrm{g\ acenafteno\ (AN)}}{100\,\mathrm{mL\ etanol\ frío}} = \mathbf{0.19\,\mathrm{g\ AN}} \;\text{queda disuelto a}\; 0^\circ\mathrm{C} $$Sin embargo, la impureza acetanilida no debería cristalizar. El cálculo siguiente muestra que 1,45 g de acetanilida pueden disolverse en el etanol frío, y dado que originalmente sólo había 0,50 g, toda la porción permanecería disuelta.
$$ 12.5\,\mathrm{mL\ etanol\ frío} \times \frac{11.6\,\mathrm{g\ AT}}{100\,\mathrm{mL\ etanol\ frío}} = \mathbf{1.45\,\mathrm{g\ AT}} \quad (\text{disueltos a }0^\circ\mathrm{C}) $$Este proceso se resume en la Figura 3. Como sólo cristaliza el acenafteno, éste puede recogerse por filtración y separarse de la impureza acetanilida en el licor madre.

La purificación de esta mezcla funciona bien en teoría porque la acetanilida es mucho más soluble (aproximadamente diez veces más) en el etanol frío que el acenafteno, por lo que puede eliminarse. En la práctica, si la cristalización se realiza con demasiada rapidez, sigue existiendo la posibilidad de que la acetanilida se incorpore al sólido en desarrollo.
Con una impureza de solubilidad similar
Si un sólido impuro contiene una impureza que tiene propiedades de solubilidad similares a las del compuesto deseado, todavía es posible purificar la mezcla mediante cristalización si la impureza está presente en una pequeña cantidad. Un ejemplo es una muestra que contiene 4,00 g de acenafteno y 0,30 g de fenantreno (el fenantreno es el 6mol% de la muestra). La solubilidad de estos compuestos en etanol se muestra en la figura 4.
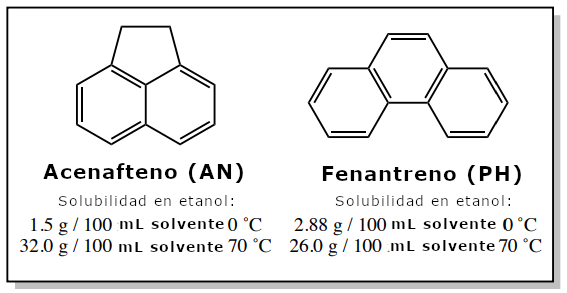
4,00 g de acenafteno puro (AN) deberían disolverse completamente en 12,5mL (véase el cálculo más abajo) de etanol casi hirviendo (el punto de ebullición del etanol es 78oC. 0,30g de fenantreno puro (PH) se disolverían en 1,2mL de etanol caliente (véase el cálculo más abajo), por lo que también se disolverían en 12,5 mL si se utilizara.
$$ 4.00\,\mathrm{g\ acenafteno\ (AN)} \times \frac{100\,\mathrm{mL\ etanol\ caliente}}{32.0\,\mathrm{g\ AN}} = \mathbf{12.5\,\mathrm{mL\ etanol\ caliente}} \;\left(70^\circ\mathrm{C}\right)\; \text{para disolver AN} $$ $$ 0.30\,\mathrm{g\ fenantreno\ (PH)} \times \frac{100\,\mathrm{mL\ etanol\ caliente}}{26.0\,\mathrm{g\ PH}} = \mathbf{1.2\,\mathrm{mL\ etanol\ caliente}} \;\left(70^\circ\mathrm{C}\right)\; \text{para disolver PH} $$Si esta solución caliente se enfría a continuación en un baño de hielo, los cálculos siguientes muestran la cantidad de cada compuesto que quedaría disuelta en el etanol frío.
$$ 12.5\,\mathrm{mL\ etanol\ frío} \times \frac{1.5\,\mathrm{g\ acenafteno\ (AN)}}{100\,\mathrm{mL\ etanol\ frío}} = \mathbf{0.19\,\mathrm{g\ AN}} \quad(\text{permanece disuelto en etanol a }0^\circ\mathrm{C}) $$ $$ 12.5\,\mathrm{mL\ etanol\ frío} \times \frac{2.88\,\mathrm{g\ fenantreno\ (PH)}}{100\,\mathrm{mL\ etanol\ frío}} = \mathbf{0.360\,\mathrm{g\ PH}} \quad(\text{permanece disuelto en etanol a }0^\circ\mathrm{C}) $$Dado que la cantidad de acenafteno es superior a la indicada, la mayor parte cristalizará (3,81 g). Sin embargo, como sólo hay 0,30 g de fenantreno, toda la porción debería permanecer en el licor madre. Los dos componentes pueden entonces separarse mediante filtración.
En esencia, las impurezas que tienen una solubilidad similar a la del compuesto de interés pueden eliminarse como impurezas solubles siempre que estén presentes en cantidades suficientemente pequeñas. Es una guía general que los sólidos que contienen menos del 5 % mol de impureza pueden ser purificados de esta manera. El proceso de cristalización esencialmente «sacrifica» una porción de cada componente al licor madre para producir un cristal puro.
Cristalización de segunda cosecha
Como se ha comentado anteriormente, una parte del compuesto de interés siempre permanece disuelta en el licor madre y se filtra. Esto no quiere decir que esta porción se pierda, ya que es posible recuperar compuesto adicional del licor madre. El disolvente puede concentrarse en el recipiente original o en un evaporador rotatorio, y puede intentarse una segunda cristalización. Una segunda cristalización a partir del licor madre de la primera cristalización se denomina «cristalización de segunda cosecha».
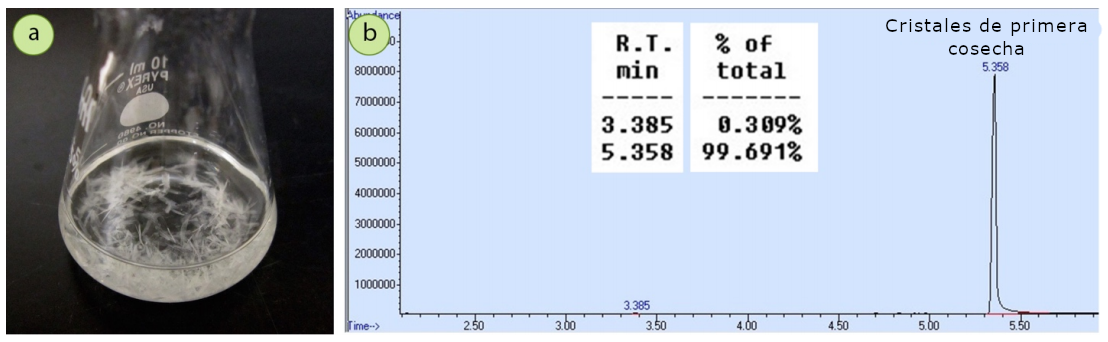
Para demostrar este proceso, una muestra de 1,16 g de ácido trans-cinámico se cristalizó a partir de un sistema de disolvente mixto metanol/agua (Figura 5). Se recogieron 0,95 g de una «primera cosecha» de cristales, lo que representa una recuperación del 82%.

Para recoger una segunda cosecha de cristales, se pipeteó el filtrado en un matraz Erlenmeyer (Figura 6a) y se hirvió para reducir el volumen a la mitad (Figuras 6b+c). Al enfriar esta solución, cristalizaron otros 0,08 g de compuesto (Figura 6d), con lo que la recuperación combinada de ácido trans-cinámico fue de 1,03 g, es decir, del 89%.

Un sólido de «segunda cosecha» debe mantenerse siempre separado de un sólido de «primera cosecha» hasta que se pueda verificar su pureza. Un cristal de segunda cosecha suele ser más impuro que un cristal de primera cosecha, ya que cristaliza a partir de una solución que contiene un mayor porcentaje de impurezas (la primera cosecha eliminó más compuesto, dejando más impurezas).
Para demostrar la pureza relativa de los cristales de la primera cosecha y de la segunda cosecha, se ha utilizado una mezcla de 1,5 g de 4-bromoacetofenona contaminada con una pequeña cantidad de acetofenona se cristalizó con etanol. Un espectro de cromatografía de la mezcla original (Figura 7b) muestra que la muestra tiene aproximadamente un 7mol% de impureza de acetofenona.
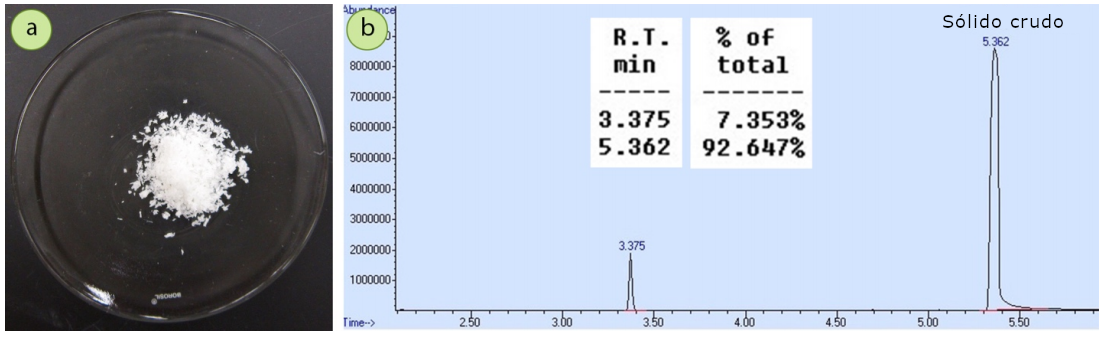
La cuantificación mediante un instrumento de cromatografía-EM puede ser a veces inexacta, por lo que la composición se calculó también mediante integraciones de H1 RMN. El espectro parcial de H1 RMN de esta muestra aparece en la figura 8, y las integraciones indican que la muestra tiene un 5,6mol% de acetofenona. Hay una buena concordancia entre los dos métodos cuantitativos.
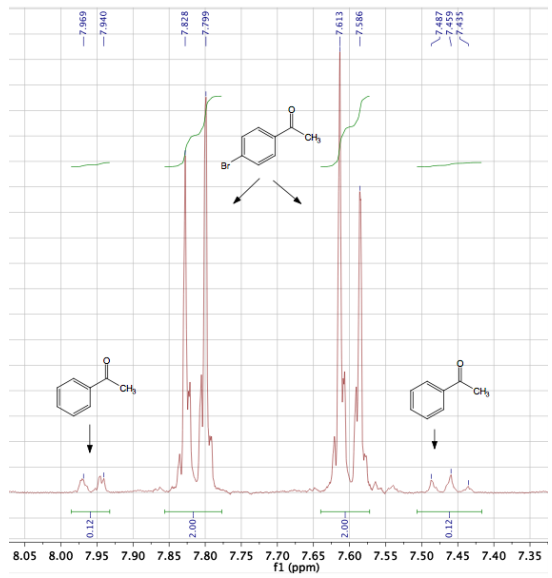
Después de cristalizar la mezcla a partir de etanol, un espectro de GC del sólido resultante (0,93 g, 63% de recuperación) mostró una drástica reducción de la impureza acetofenona, hasta cantidades casi insignificantes (Figura 9b). La cristalización purificó muy bien la 4-bromoacetofenona.
El filtrado de la primera cristalización se hirvió para reducir el nivel de disolvente, y se obtuvo una segunda cosecha de cristales (0,18 g, recuperación combinada del 77%). Los cristales de la segunda cosecha tenían un notable tinte amarillo, mientras que los de la primera cosecha eran de color blanco puro. Un espectro de cromatografía de los cristales de la segunda cosecha (Figura 10b) mostró que la acetofenona se había incorporado un poco al sólido cristalino, demostrando que los cristales de la segunda cosecha son menos puros que los de la primera.


La recuperación de una segunda cosecha de cristales no se suele hacer en los entornos de pregrado, ya que la recuperación adicional de una segunda cosecha suele ser modesta (Figura 11). Sin embargo, el proceso puede realizarse si el material es bastante caro o si queda una gran cantidad disuelta en el licor madre. Por esta última razón, es una buena idea guardar el licor madre hasta que se pueda calcular el rendimiento. Si la recuperación es bastante baja, podría intentarse una segunda cristalización.
Para más información Quantitating Crystallization



